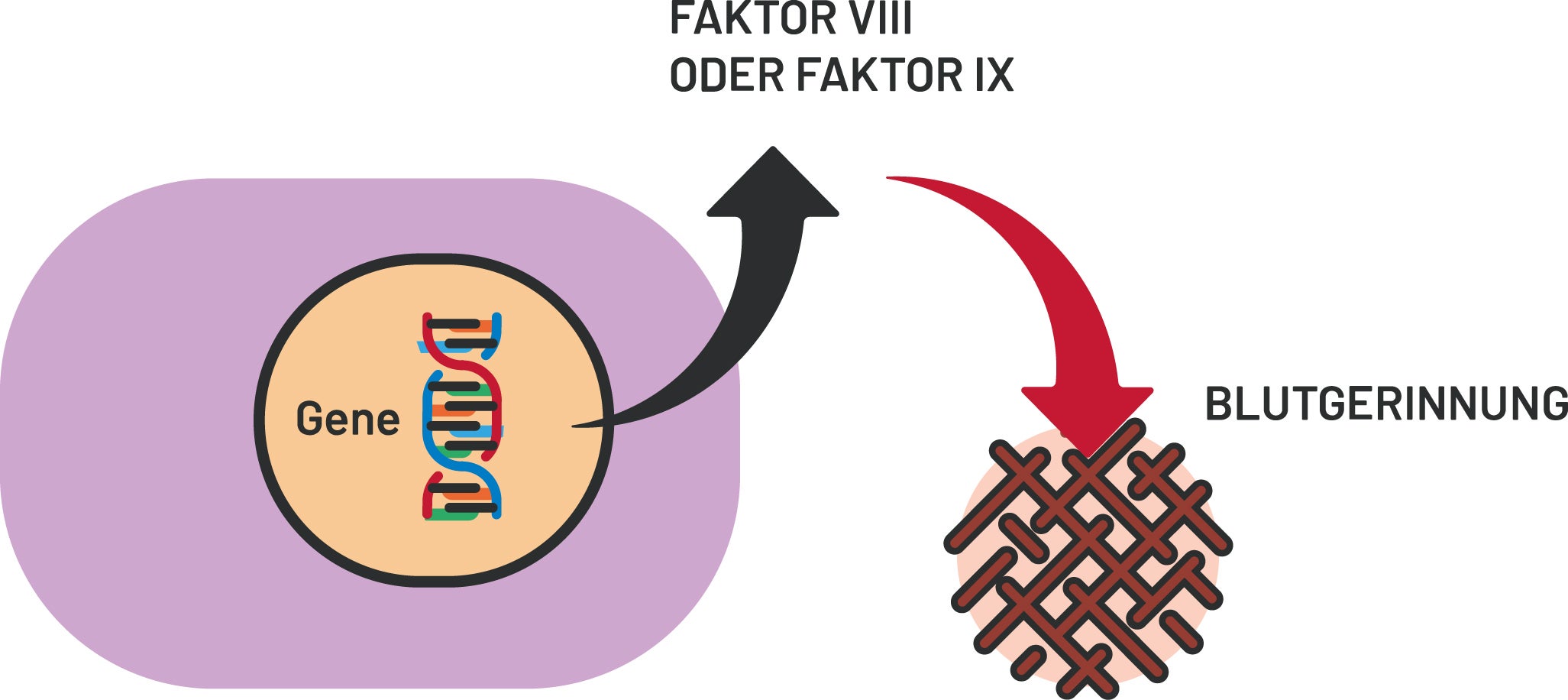
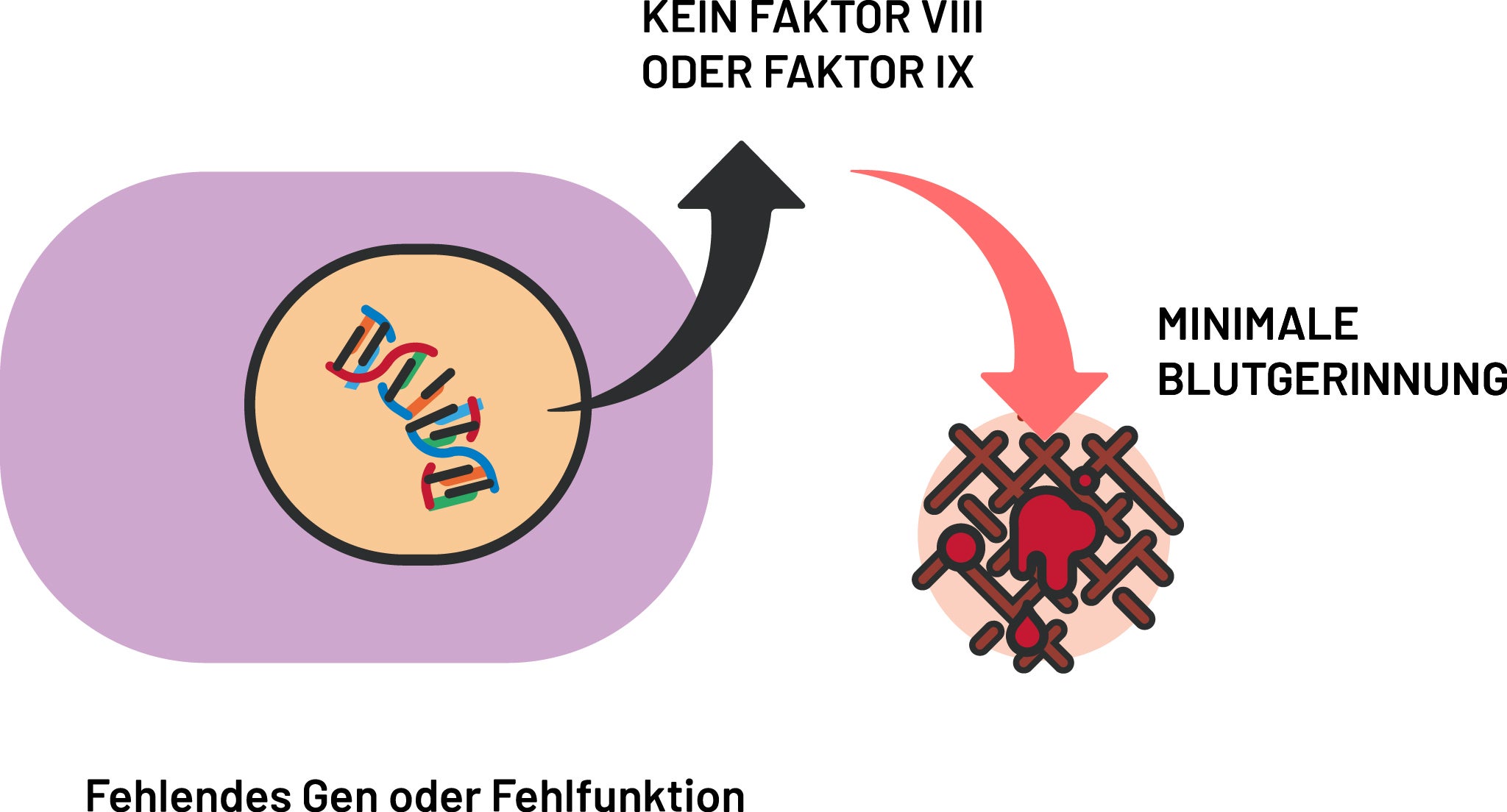
Hämophilie
Copyright Dres. Schlegel + Schmidt Med. Kommunikation GmbH
Ausgangssituation 1
Die Mutter hat ein defektes X-Chromosom und ist somit Konduktorin. Der Vater ist gesund. Mit einer Wahrscheinlichkeit von 50 % werden die Töchter Konduktorinnen der Krankheit sein. Die Söhne werden mit einer Wahrscheinlichkeit von 50 % an Hämophilie erkranken. (Abb. 1)
Ausgangssituation 2
Der Vater hat ein defektes X-Chromosom und ist daher an Hämophilie erkrankt. Die Mutter ist gesund und hat zwei intakte X-Chromosomen. Alle Töchter werden folglich zu Konduktorinnen. Alle Söhne werden gesund sein. (Abb. 2)
Ausgangssituation 3
Der Vater hat ein defektes X-Chromosom und ist daher an Hämophilie erkrankt. Die Mutter ist Konduktorin und hat ebenfalls ein defektes X-Chromosom. Die Töchter werden mit 50 % Wahrscheinlichkeit an Hämophilie erkranken – in jedem Fall werden sie zu Konduktorinnen. Die Söhne werden mit einer Wahrscheinlichkeit von 50 % an Hämophilie erkranken.
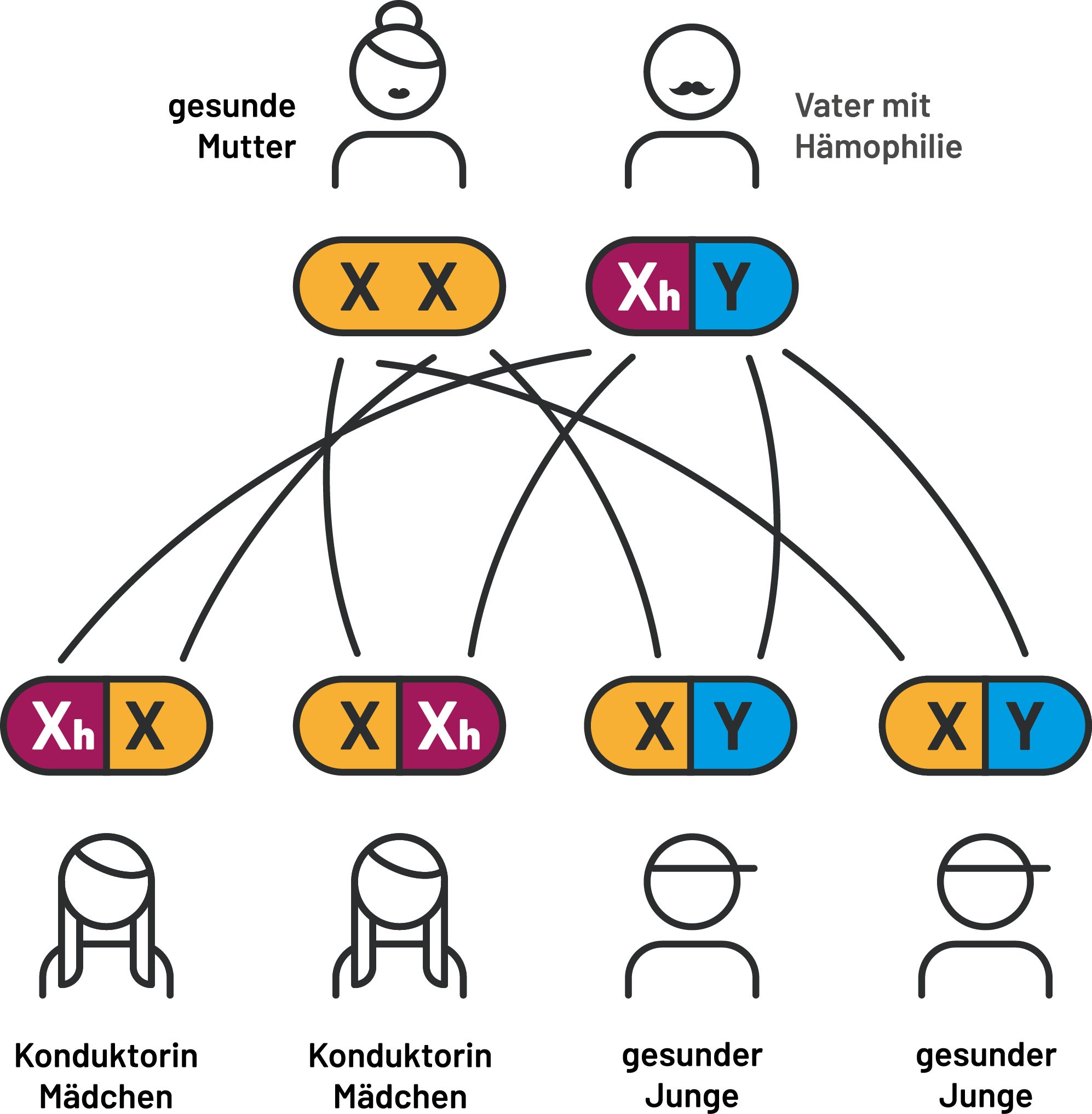
Abb. 1
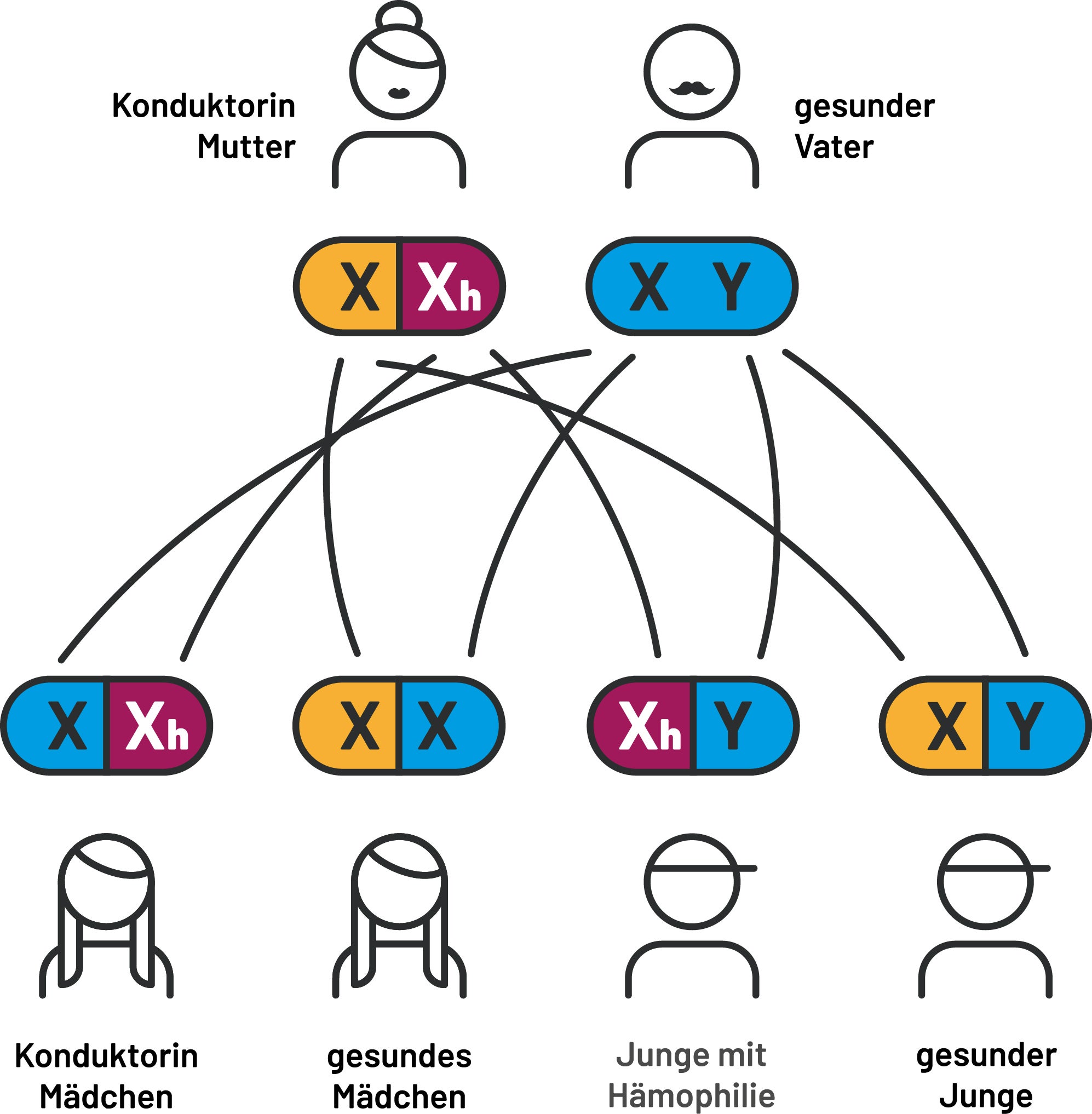
Abb. 2
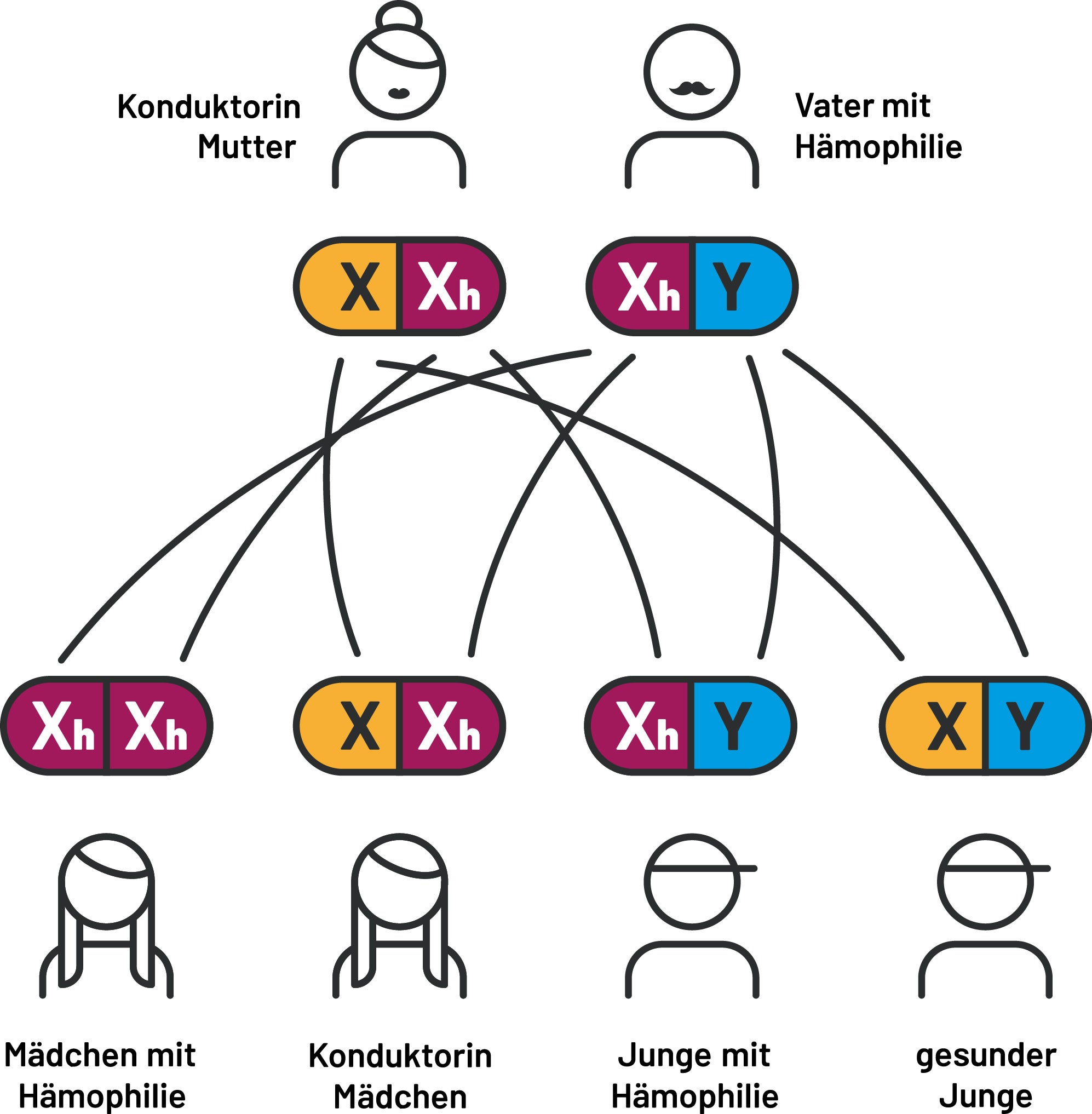
Abb. 3
Erweiterte Diagnostik
Initial wird die Aktivität der Gerinnungsfaktoren einschließlich der Thrombozytenzahl, Prothrombinzeit und partiellen Thromboplastinzeit gemessen sowie die Quantität und Funktionalität von FVIII und FIX analysiert. Bei Bedarf werden weitere Faktoren untersucht.7 Ein gezielter Gentest kann – bei schwangeren Konduktorinnen auch pränatal – die Diagnose sichern.1
Weitere Informationen zur spezifischen Labordiagnostik finden Sie hier.

Schwangerschaft und Geburt
Konduktorinnen mit Kinderwunsch können sich im Hämophilie-Zentrum beraten lassen. Damit Schwangerschaft und Geburt sicher für Mutter und Baby verlaufen, sollten alle Behandelnden wie Hämostaseolog:innen, Frauenärzt:innen und Hebammen informiert sein. Wurde beim ungeborenen Kind eine schwere Hämophilie diagnostiziert, erfolgt die Entbindung am besten in einer Klinik, die über Erfahrung bei der Geburt von Kindern mit Hämophilie und ggf. Betreuung von Müttern mit Blutgerinnungsstörungen verfügt.1
Weitere therapeutische Maßnahmen sind von der individuellen Situation der Betroffenen abhängig. Mehr dazu finden Sie hier.

EXA/DE/HG/0281